




Para este estudio se generaron un total de 192 nuevos genomas de SARS-CoV-2-Italian. El historial de viajes estuvo disponible para 137 (71,3%) pacientes. Todos informaron no haber viajado internacionalmente en las dos semanas anteriores al inicio de los síntomas. Se informó de un caso de contacto con un viajero de Bangladesh. La información clave del paciente se informa en la Tabla 1.
Análisis de conjuntos de datos italianos
Diversidad genómica basada en clasificación de linaje/rama
Las cepas más prevalentes fueron B.1 (n = 222, 47,7 %, incluidas 32 cepas derivadas de B.1 como B.1.76, B.1.91, B.1.104, B.1.142, B.1.153, B.1.177) ., b 1.179, b 1.222, b 1.225, b 1.356, b 1.610) y b 1.1 (n = 141, 30.3%, incluyendo 19 cepas derivadas de b 1.1 como b 1.1.28, b. 1.1.61 y B. 1.1.161, B.1.1.202, B.1.1.232, B.1.1.331 y B.1.1.372) seguido de las cepas B (n = 73, 15,7 %) y B. 1.1.1 (n = 29 , 6,2%). La siguiente clasificación de diapositivas mostró una alta prevalencia de los bloques 20A (n = 207, 44,5 %) y 20b (n = 141, 30,3 %), seguidos por 19a (n = 84, 18,1 %) y 20d (n = 29, 6,2 %). . %). Solo 4 cepas fueron Clyde 20 c (0,9%).
La distribución geográfica de las cepas/cepas del SARS-CoV-2 en Italia (Fig. 1) mostró varios patrones epidemiológicos diferentes. Algunas áreas, principalmente en el centro-norte de Italia (Friuli-Venezia Giulia, Marche, Emilia-Romagna, Lombardy, Lazio) mostraron una alta prevalencia de B.1/20A (entre 70 y 100%). Otras regiones, especialmente en el centro y sur de Italia (Cerdeña, Sicilia, Abruzzo, Apulia) registraron la mayor prevalencia de B.1.1/20B (del 57 % a más del 90 %). Otras regiones mostraron una proporción igual de ambas cepas (Basilicata, Liguria, Toscana, Umbría). Dos regiones tenían un patrón único: Veneto, donde el linaje más prevalente era B/19A (66/97, 68 %) y Piamonte, donde el 73 % (27/37) mostraba un linaje B.1.1.1/20D.
Distribución espacial de linajes y masas. (aY el B(Mapa de Italia que muestra la distribución de proporciones)a) y asignación de clados (B) en cada región.
Se observó un cambio en la prevalencia de los linajes SARS-CoV-2 entre febrero y mayo. Las cepas más detectadas fueron B/19A y B.1/20A en febrero y la primera quincena de marzo, representando el 88% de todos los genomas obtenidos en ese periodo de tiempo. Luego, a partir de la segunda quincena de marzo, B.1.1/20B y otros linajes (B.1.1.1/20D) se hicieron más frecuentes (60,7% entre el 15 y el 31 de marzo, 46,2% en abril, 51,6% en mayo).
Distancias genéticas y análisis mutacional
La distancia media general p entre todos los aislamientos italianos fue de 3,9 (SE: 0,4) s/10000 nts que corresponde a una media de 10,1 (SE: 1,01) sustituciones por genoma. La distancia genética se mantuvo pequeña con una media de 10,23 (SE: 1,09) de las variantes, de las cuales 3,13 (SE: 0,59) eran sinónimas y 6,85 (SE: 0,79) no sinónimas. Se observó una mayor heterogeneidad en las secuencias de Piamonte (20,4, SE: 1,6) y Sicilia (18,4, SE: 1,2) en comparación con las otras regiones. Curiosamente, se ha registrado un número creciente de diferencias a lo largo del tiempo, desde 5,7 (SE: 0,81) en febrero hasta 20,1 (SE: 1,1) en mayo.
Diecisiete sustituciones de aminoácidos estaban presentes en más del 10 % de los aislados italianos, pero solo una de ellas estaba en la proteína de la espiga (D614G). No se observaron mutaciones en el dominio de unión al receptor (RBD) en todo el conjunto de datos de secuenciación italiano. Solo once secuencias de la cepa B en el conjunto de datos completo, todas de Veneto (clado 19A), portaban T1543I en orf1a. En general, las secuencias B mostraron un patrón mutacional distinto al de otras cepas, incluidas las mutaciones L3606F y G251V en orf1a y orf3a, respectivamente. El linaje B.1.1.1 presentó sustituciones adicionales en comparación con las cepas B.1 y B.1.1 como T1246I en orf1a en todos los aislados. La Tabla 2 muestra las sustituciones de aminoácidos más comunes desglosadas por proporción y masa.
Análisis filogenético por ML y métodos bayesianos
Un análisis filogenético bayesiano asignando cada punta a su linaje mostró 4 grandes hendiduras de gran importancia correspondientes a los principales linajes que circulan en Italia (b, b1, b1.1, b1.1.1) (Fig. 2). B1, B.1.1 y B.1.1.1 se fusionaron entre sí, mientras que B se separó de forma independiente. Las secuencias chinas tienden a separarse en el conjunto exterior de bloques italianos dentro de los linajes B y B.1. La estimación de tMRCA para masas mayores indica que la cepa B se propagó a Italia en la última semana de enero de 2020, la cepa B.1.1 surgió más tarde, a mediados de febrero, y la B.1.1.1 fue la más reciente, propagándose a principios de marzo. El análisis de ML mostró tMRCA similares pero con períodos encubiertos más amplios (Tabla 3).
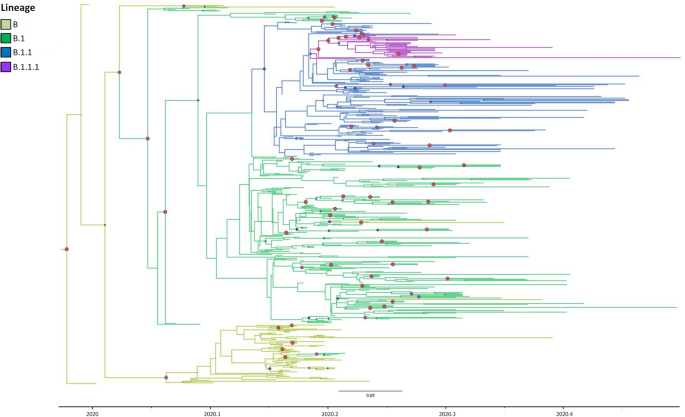
SARS-CoV-2 Árbol geográfico de cepas bayesianas de 479 cepas. Los círculos grandes de color rojo y violeta indican la probabilidad posterior más alta que va de 1 a 0,9. Las ramas se colorean según las proporciones más probables de los nodos descendentes.
Geografía en Italia
El geógrafo del SARS-CoV-2 identificó a China como el sitio de la raíz del árbol (Fig. 3 y Fig. 1 complementaria). Se han identificado cuatro grandes grupos principales. Los primeros grupos en Lombardía y Véneto estaban directamente relacionados con China, mientras que otros aparecieron más tarde (hacia la segunda quincena de marzo) en Abruzos y Piamonte. Al combinar la geografía con las cepas del SARS-CoV-2, las reconstrucciones del estado ancestral mostraron que los linajes B y B.1 se extendieron desde China hasta Véneto y Lombardía, respectivamente. Mientras que el linaje B aparentemente permaneció confinado a Veneto (y fue extinguido con éxito), el linaje B.1 se extendió desde Lombardía a otras regiones italianas (Veneto, Emilia-Romagna, Abruzzo, Marche, Puglia, Friuli Venezia Giulia y Lazio). El linaje se extiende por 1,1 desde el centro de Italia (Abruzzo) a otras regiones italianas (Veneto, Lombardía, Puglia, Cerdeña). Finalmente, el linaje B.1.1.1 apareció más tarde y aparentemente permaneció localizado en Piamonte sin extenderse más a otras regiones.
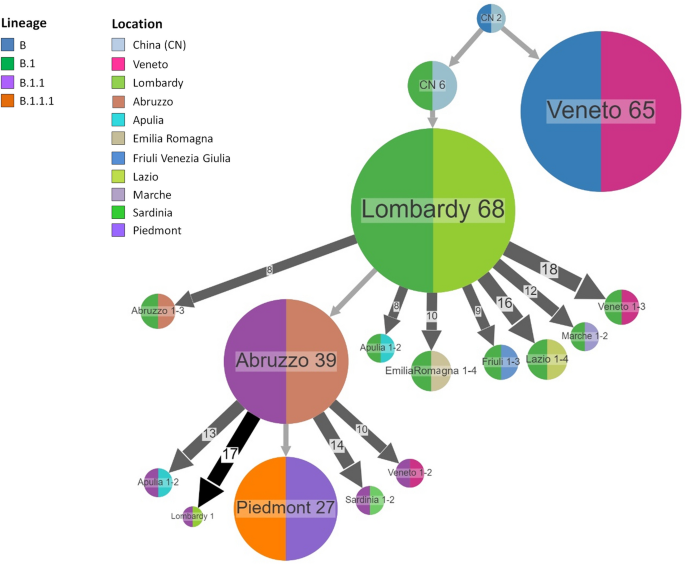
Reconstrucción ancestral de cepas de SARS-CoV-2 B.1 utilizando el conjunto de datos italiano. La figura muestra las visualizaciones comprimidas producidas por PastML usando la aproximación de probabilidad marginal posterior (MPPA) con un modelo similar a F81. Diferentes colores corresponden a diferentes regiones geográficas y linajes italianos. Los números dentro (o al lado) de los círculos indican el número de linajes asignados al nodo dado.
Análisis de conjuntos de datos internacionales
bloques italianos
El análisis filogenético por ML de todo el conjunto de datos, incluidos los genomas italiano, europeo y chino, mostró que la mayoría de los aislamientos italianos estaban dispersos en todo el árbol. Se incluyeron un total de 80 (de 465, 17,2%) aislados italianos en 22 grupos de gran apoyo (Tabla 4). De estos, 12 (54,5%) estaban dentro de la cepa B.1, cinco (22,7%) eran B.1.1/20B, tres (13,6%) eran B.1.1/20D y dos (9,1%) eran B/19a. Todos menos uno de los ensamblajes B.1 se clasificaron como clado 20A. El grupo 19 fue la única excepción e incluyó cuatro cepas italianas clasificadas como clado 20C (todas de Roma), que mostraron una caída media de tMRCA en marzo de 2020. Tres grupos (13,6 %) eran únicos (incluido solo un aislado italiano). secuencias), que probablemente corresponden a introducciones esporádicas seguidas de circulación limitada, mientras que los diecinueve grupos restantes incluyen al menos dos aislamientos italianos, lo que sugiere transmisión local. Trece de ellos (68,4%) contenían únicamente cepas italianas (lo que indica una propagación local de esta cepa), mientras que 6 (31,6%) incluían aislados de otros países europeos, y uno de ellos (B1) también incluía un solo genoma chino.
La estimación de los grupos tMRCA por el método ML confirmó que los primeros eventos de transmisión en Italia datan de alrededor de la segunda quincena de enero y principios de febrero. Dieciocho grupos tenían un ancestro común anterior a las medidas de confinamiento en nuestro país. En particular, los grupos B.1/20A (10/14) predominaron en puntos de tiempo anteriores (antes de marzo) mientras que predominaron en marzo (20B, 20 C y 20 D) (6/8). Además, los grupos mixtos y únicos prevalecieron inicialmente, mientras que los grupos italianos puros fueron los únicos observados después del cierre. El grupo más antiguo (n.º 1), el pedigrí era B.1/20A, con una fecha promedio del 20 de enero de 2020 (IC95% 01/08-24/01/2020) e incluía solo cuatro subespecies del norte de Italia: una de Lodi, dos de Milán (los lugares donde se identificaron por primera vez los casos originales de COVID-19 en Italia) y uno de Piacenza. El primer grupo B.1.1 estaba fechado el 10/02/2020 (IC95% 01/28/2020-03/12/2020) e incluía 3 aislados italianos de Abruzzo. Tres clústeres B.1.1.1/20D de fecha 2 de marzo (IC95% 22/02/2020–02/03/2020). Solo se observaron dos pequeños grupos italianos sostenidos por grandes cinturones cebadores dentro del árbol ML, incluidos los aislamientos B/19A. En particular, un grupo italiano puro contenía 11 genomas de Veneto (provincia de Padova), caracterizado por la sustitución T1543I en orf1a, y no se detectó en ninguno de los otros genomas B/19A en nuestro conjunto de datos internacional.
Análisis filográfico en Europa
Al incorporar la reconstrucción del país ancestral del sitio con el linaje (Fig. 4 y Fig. 2 complementaria), los análisis mostraron que B.1 probablemente se originó en China y se extendió a varios países europeos llegando a Italia varias veces, formando un gran grupo que inicialmente incluyó 59 (alrededor de la primera semana de marzo) y finalmente 198 genomas, y otras 6 introducciones independientes que corresponden principalmente a un grupo de genomas caracterizados solo por la sustitución D614G pero carente de otras sustituciones, en particular P314L en el RdRp que define el clado 20A (linaje B1, clado 19a).
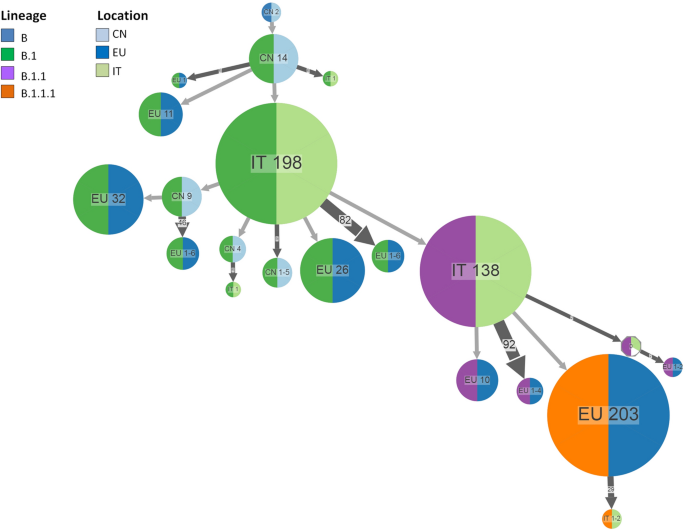
Reconstrucción ancestral de cepas de SARS-CoV-2 B.1 utilizando el conjunto de datos europeo. La figura muestra las visualizaciones comprimidas producidas por PastML usando la aproximación de probabilidad marginal posterior (MPPA) con un modelo similar a F81. Diferentes colores corresponden a diferentes países y linajes europeos. Los números dentro (o al lado) de los círculos indican el número de linajes asignados al nodo dado. Se muestran las predicciones del escenario del ancestro común (común) y la Predicción lateral máxima (MAP) para nodos inciertos (que se muestran como iconos octogonales). CN, China; TI, Italia, UE, Europa.
Comenzando en Italia, el B1/20A se extendió a otros países europeos y luego se reintrodujo en China. Un segundo gran grupo italiano, que incluye 138 genomas del linaje B.1.1, surgió del grupo italiano B.1. Se han observado múltiples introducciones de B.1.1 desde Italia a otros países europeos. Un gran grupo (n = 203 genomas) correspondiente a la cepa B.1.1.1 apareció en Europa a principios de marzo y llegó a Italia más tarde (segunda quincena de marzo) (Fig. 4). Un total de 7 nodos permanecieron sin especificar. Un análisis separado realizado para distinguir entre países europeos (en lugar de considerar un solo grupo generalizado) generalmente confirmó este escenario y permitió reconstruir la propagación de la epidemia en los países europeos con más detalle (Figura 3 complementaria).
El análisis del linaje B mostró que solo dos nodos permanecieron sin identificar entre Europa y China (Fig. 4 complementaria). La visualización (Fig. 5) sugirió varias introducciones de China a Italia a partir de finales de febrero. Se observó un grupo correspondiente al grupo #5 descrito anteriormente, mientras que las otras cepas aparentemente representan múltiples introducciones independientes que forman pequeños grupos de no más de dos secuencias. También se observaron introducciones esporádicas de Europa. En contraste con la reconstrucción ancestral del linaje B.1, este escenario era diferente ya que los flujos migratorios parecían detenerse en Italia sin extenderse más.
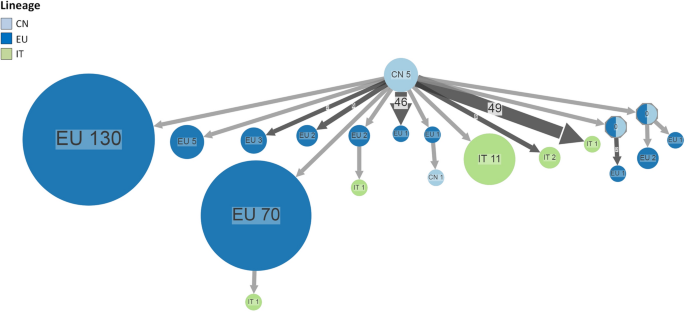
Reconstrucción de ancestros de SARS-CoV-2 B utilizando el conjunto de datos europeo. La figura muestra las visualizaciones comprimidas producidas por PastML usando la aproximación de probabilidad marginal posterior (MPPA) con un modelo similar a F81. Diferentes colores corresponden a diferentes países y linajes europeos. Los números dentro (o al lado) de los círculos indican el número de linajes asignados al nodo dado. Se muestran las predicciones del escenario del ancestro común (común) y la Predicción lateral máxima (MAP) para nodos inciertos (que se muestran como iconos octogonales). CN, China; TI, Italia, UE, Europa.
El análisis intereuropeo (Figura 5 complementaria) destacó el mismo escenario ancestral pero no mostró ninguna introducción de Europa.

«Propenso a ataques de apatía. Explorador de aspirantes. Analista ávido. Fanático de Internet. Comunicador»
More Stories
Probando nueva tecnología de mapeo 3D para transformar la exploración espacial y beneficiar a las industrias de la Tierra
¿Júpiter tiene anillos? Sí, lo es
El efecto de la dieta sobre las bacterias intestinales proporciona nuevas pistas en el tratamiento de la enfermedad de Parkinson