





En un estudio reciente publicado en bioRxiv* El equipo de preimpresión, un equipo de investigadores de Washington, Estados Unidos, investigó la transmisión de la diversidad genética viral durante el brote del coronavirus 2 (SARS-CoV-2) que se propagó ampliamente entre los miembros de la tripulación en un barco de pesca.
El SARS-CoV-2 resultó en infecciones breves y autolimitadas con desarrollo viral durante varias rondas sucesivas de infección, cada una interrumpida por un cuello de botella en la transmisión. Los eventos generalizados juegan un papel importante en la transmisión del SARS-CoV-2. Dichos eventos incluyeron condiciones altamente favorables para la transmisión viral y demostraron diferentes patrones de evolución viral.
estancia: Estrechos cuellos de botella de transmisión y diversidad viral limitada dentro del huésped durante un brote de SARS-CoV-2 en un barco de pesca. Haber de imagen: Natalia Dobrianskaya/Shutterstock
diseño del estudio
El presente trabajo de investigación extrajo ARN de hisopos nasales de miembros del equipo positivo para SARS-CoV-2 y se generaron bibliotecas de secuenciación primaria. Con valores de umbral de ciclo de reacción en cadena de polimerasa de transcripción inversa cuantitativa (RT-qPCR) por debajo de 20, los investigadores volvieron a secuenciar muestras de las bibliotecas originales amplificando el ARN viral a través de PCR. Los investigadores obtuvieron un promedio de 1.113.690 lecturas de mapas de cada biblioteca.
La canalización Snakemake procesó los datos de los archivos de secuencia sin procesar. Las lecturas para SARS-CoV-2 se seleccionaron de archivos FASTQ usando 31 bases que coincidían con la referencia genética Wuhan-1/2019 usando Descontaminación de Bestus Bioinformaticus usando Kmers (BBDuk). Se verificó la calidad de los archivos del mapa de alineación bialineada (BAM) utilizando las herramientas del mapa de alineación de secuencia (SAM).
Para el análisis de secuencias, los investigadores recolectaron los genomas acordados y los organizaron en alineaciones múltiples utilizando la transformada rápida de Fourier (MAFFT). Usando genomas alineados, se construyó un árbol IQ TREE con 1000 repeticiones. Para incluir genomas similares que estaban en el barco, se generó en Washington una base de datos de todas las secuencias de la Herramienta básica de búsqueda de alineación local (BLAST) durante y antes del brote.
Finalmente, las variables se definieron utilizando un Python Script personalizado. Se identificaron polimorfismos de un solo nucleótido (SNP), y se registró y anotó el número total de ocurrencia de SNP para efectos de codificación. Los investigadores también utilizaron tres programas de comunicación diferentes: ivar, varscan2 y lofreq.
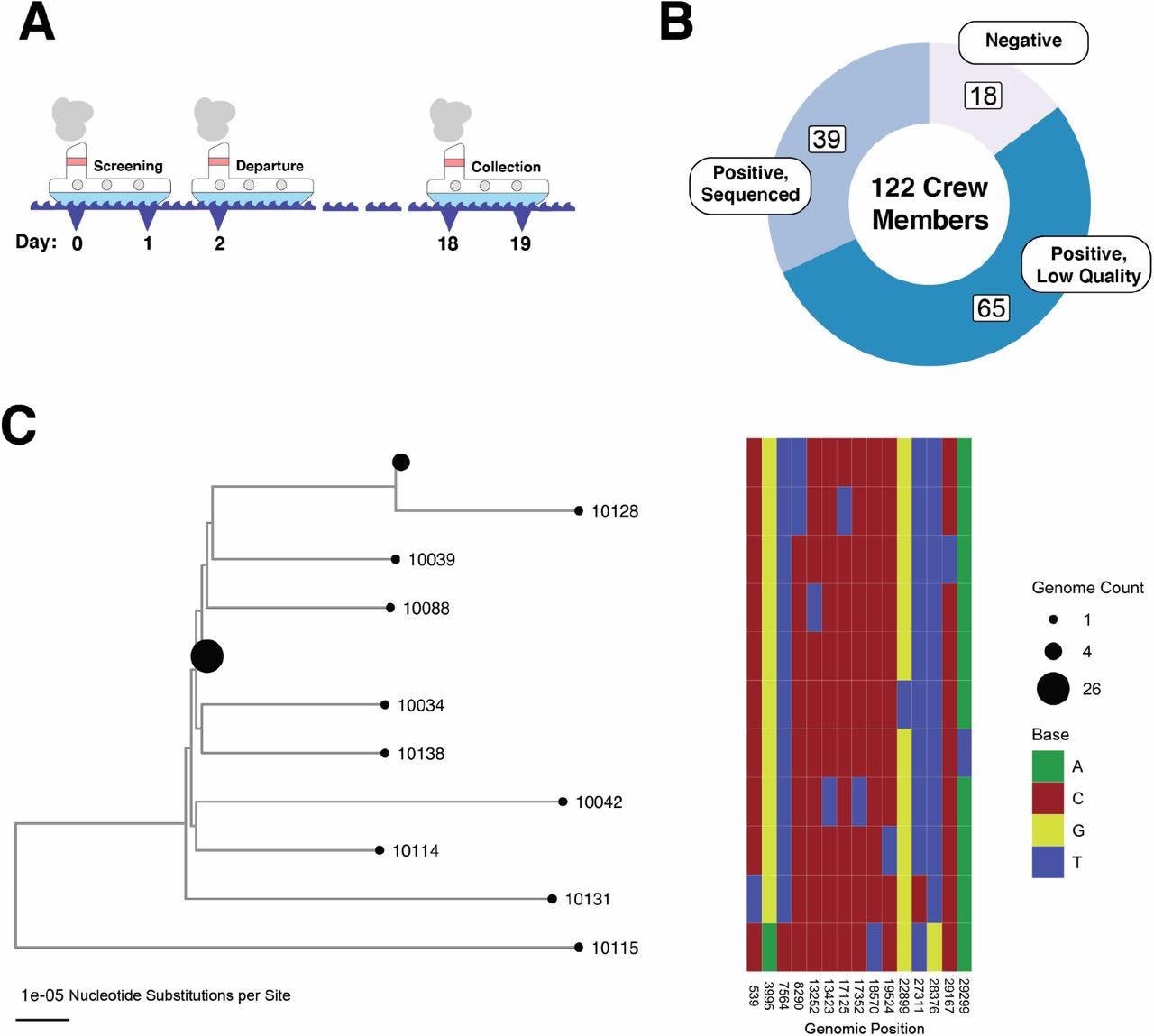
Un brote de SARS-CoV-2 en un barco de pesca aislado es un grupo de infecciones vinculadas a un vínculo epidémico. (a) Un diagrama esquemático que muestre la cronología del brote del barco pesquero. Todas las muestras utilizadas en este estudio se tomaron el día 18 como se muestra en la figura (en relación con el inicio del ensayo previo a la partida). (b) Un gráfico de anillos que muestra la distribución de muestras para los 122 miembros de la tripulación. (c) La evolución del genoma del SARS-CoV-2 desde el barco. El mapa de calor a la derecha muestra las diferencias de nucleótidos entre los genomas del árbol. Los números de identificación de muestra para las muestras de los miembros de la tripulación designan los nodos de hoja del árbol, excepto aquellos nodos que contienen más de un genoma idéntico. Los tamaños de los nodos son proporcionales al número de secuencias: hay nodos que representan 26 secuencias idénticas (10101, 10126, 10133, 10105, 10108, 10130, 10031, 10110, 10030, 10124, 10029, 10102, 10038, 10094, 100127 10117, 10106, 10091, 10093, 10127, 10116, 10040, 10090, 10036, 10089) y nudos que representan 4 secuencias idénticas (10107, 10129, 10113, 10028); Todos los demás nodos representan secuencias únicas.
los resultados
Los resultados mostraron que durante la evaluación previa a la partida de 120 personas de Seattle, ninguna de ellas dio positivo por SARS-CoV-2. Sin embargo, después de abordar el barco, el 80% de la tripulación dio positivo por SARS-CoV-2, lo que indica una tasa de ataque secundario más alta. Curiosamente, antes de abandonar el barco, los anticuerpos neutralizantes estaban presentes en solo tres de los miembros de la tripulación, y ninguno de los individuos cumplía con la definición de infección.
Entre las muestras de personas que dieron positivo por SARS-CoV-2 después de que el bote fuera devuelto a la costa, 39 muestras de hisopos nasales tenían niveles suficientemente altos de ARN viral (valor Ct <26) que se utilizaron para ensamblar secuencias virales a partir de datos de secuenciación profunda. El equipo notó que el 75% de las secuencias virales ensambladas desde el bote eran similares a al menos otra secuencia que representaba un evento generalizado.
Durante la secuenciación ultraprofunda de las muestras, de las 39 muestras anteriores, 23 mostraron suficiente ARN viral (valor de Ct inferior a 20) para la secuenciación metagenómica. Los investigadores utilizaron un límite estricto en la profundidad de secuenciación considerando secuencias con más del 80 % del genoma oculto con 100 lecturas en una o más iteraciones mientras realizaban el análisis final. El equipo no observó ningún sesgo de longitud del genoma viral en la cobertura de la secuencia y señaló que los resultados obtenidos eran sólidos para el uso de otros métodos de comunicación de variantes.
Los investigadores observaron que dentro de cada miembro de la tripulación, la diversidad del grupo de virus era limitada, con un promedio de tres variantes dentro del huésped por miembro con una frecuencia relativamente baja, con solo unas pocas <10% apareciendo. No hubo asociación entre los valores de hisopado nasal y el número específico de SNP. Además, no se observó ningún patrón reconocible en el sitio del polimorfismo en el genoma viral.
Los investigadores notaron, mientras identificaban mutaciones, que la mayoría de las variantes de baja frecuencia eran específicas o fijadas a un solo individuo, y en frecuencias intermedias, no se observaron las variantes estables. En el barco se identificaron cuatro alelos de baja frecuencia en varios individuos —A4229C, C9502T, G14335T y T18402A— con una frecuencia no superior al 5 %.
La variante C9502T estaba presente en un tramo de homopolímero de timina, una asociación conocida de llamadas pseudovariantes en los datos de secuenciación del SARS-CoV-2. Además, G14335T y A4229C mostraron un sesgo posicional en las lecturas alineadas. Finalmente, T18402A mostró una diferencia de frecuencia significativa entre las réplicas. Todas estas características de los alelos variantes comunes de baja frecuencia sugirieron que estos alelos son técnicamente artefactos de secuencia en lugar de verdaderas mutaciones.
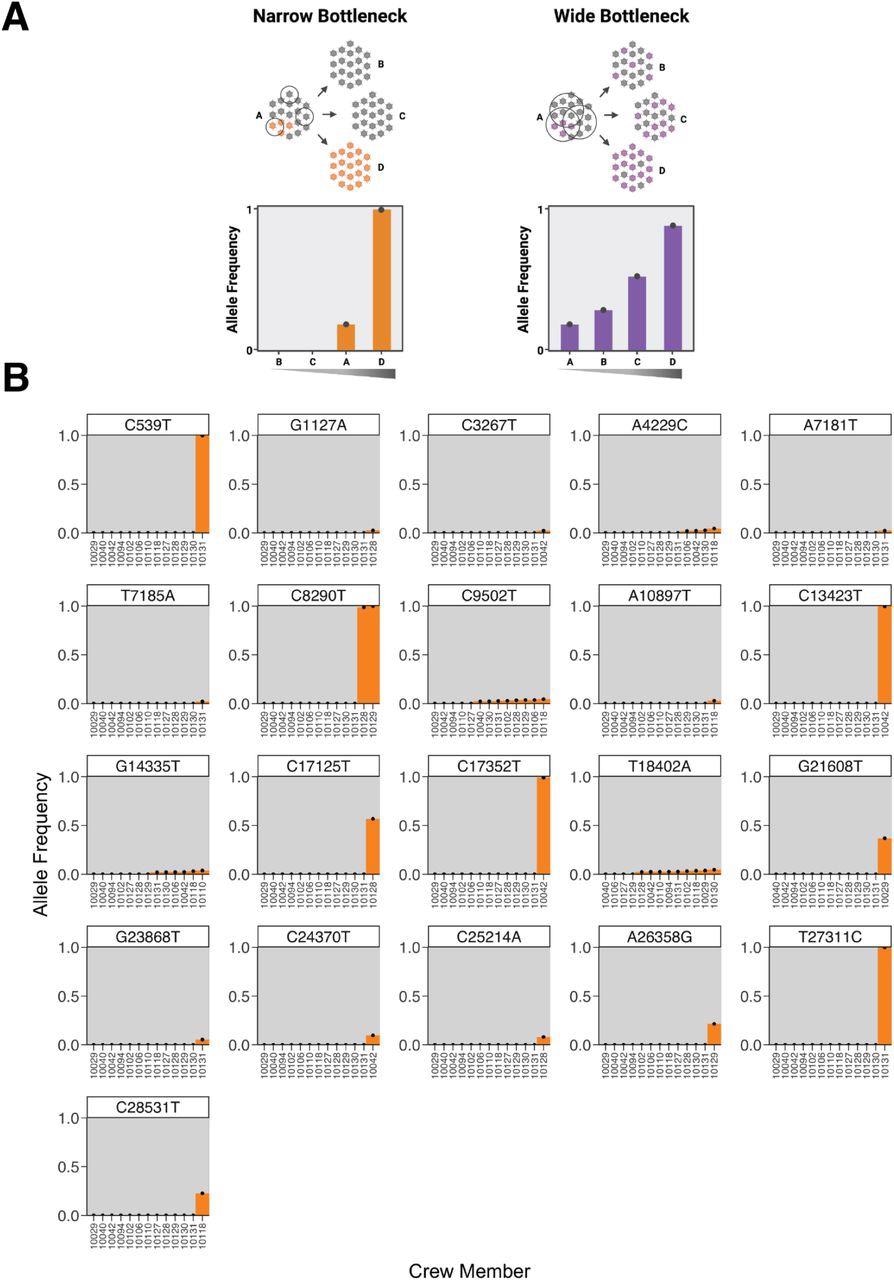
El espectro de ligera variación común indica que el cuello de botella de la transmisión es estrecho. (a) Una gráfica que muestra el patrón esperado de las frecuencias alélicas observadas para las covariables en un escenario estrecho o amplio. (B) Cada gráfico representa la frecuencia de un polimorfismo de un solo nucleótido (SNP) entre los miembros de la tripulación. Las variantes se denominan relativas a la secuencia ancestral del virus introducido en el barco según se infiere de la cepa de los genomas de los tripulantes. El eje x está dispuesto de acuerdo con la frecuencia variable.
conclusión
Esta investigación demostró que en un evento súper alto de un brote de SARS-CoV-2 en un barco de pesca, las personas relacionadas epidemiológicamente en el barco compartieron poca o ninguna diversidad viral. Además, la evolución del genoma viral ha implicado la estabilización de mutaciones a una frecuencia baja ocasional en lugar del mantenimiento continuo de la diversidad viral dentro del huésped.
Las observaciones del trabajo de investigación actual sugirieron que los cuellos de botella de transmisión estrechos son una característica global de la transmisión del SARS-CoV-2. Sin embargo, el estudio actual tiene algunas limitaciones. Por ejemplo, se obtuvieron datos de secuencia para un pequeño número de miembros de la tripulación de un barco (13 de 122) y se subcuantificó la cuantificación del cuello de botella del transporte.
*Nota IMPORTANTE
bioRxiv Publica informes científicos preliminares que no han sido revisados por pares y, por lo tanto, no deben considerarse concluyentes, orientar la práctica clínica o el comportamiento relacionado con la salud ni tratarse como información establecida.

«Propenso a ataques de apatía. Explorador de aspirantes. Analista ávido. Fanático de Internet. Comunicador»
More Stories
Un fenómeno parecido a un arco iris puede brillar en el infernal exoplaneta WASP-76b
El caos cósmico y las pistas en rápidas ráfagas de radio
SpaceX lanzará satélites Starlink en la misión número 40 de la compañía en 2024